

Helping FDA Help Itself: Voluntary Submissions of Allegations of Regulatory Misconduct
The FDA Law Blog
JUNE 27, 2023
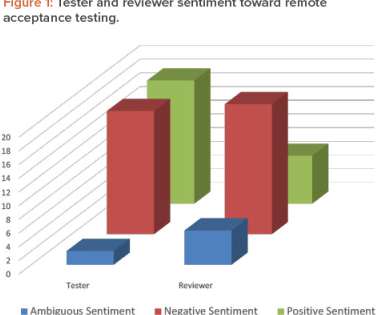
Walsh — Last fall, we blogged about the process FDA uses to review allegations of regulatory misconduct against device manufacturers, suggesting greater transparency on the FDA process was needed (see here ).


































Let's personalize your content