This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The new research analyzed paperwork submitted for FDA approval to market a device for 767 oximeters that had been approved between 1978-2024 and had accessible information about performance testing.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents.
Having all of these details in accessible, printed documents keeps patients informed and mitigates the risk of medication errors. However, there’s a growing movement across the world to eliminate printed medication information, making it harder for patients to learn about the drugs that, in many cases, save their lives.
BrainStorm filed for approval over the FDA’s protest, insisting that NurOwn has demonstrated benefits for a subset of ALS patients with milder disease.
The US FDA’s Center for Devices and Radiological Health (CDRH) dropped a bevy of new digital health guidances and reports today and yesterday, providing some long-awaited clarity and peeks into the agency’s future plans. . The push started yesterday with the 31-page key findings report from the FDA Pre-Certification Program pilot.
Walsh — Among FDA-regulated establishments and stakeholders, there is one word that makes everyone go on edge – the dreaded FDA “inspection.” In FY2023, FDA conducted over 1000 inspections under the BIMO program. We also detail some of our recommended best practices to achieve success when FDA comes knocking.
Draft guidance published by the US Food and Drug Administration (FDA) in December 2023, discussed quality considerations for topical ophthalmic drug products, including key considerations for extractables and leachables (E&L) testing. This document is revised from a version published in October 2023.
The FDA has published draft guidance on how digital health technologies (DHTs) like smart and wearable devices can be used to capture data remotely from patients in clinical trials – an approach that has come to the fore since the start of the pandemic. The draft is open for comment until 22 March.
The list of AI devices was first published after a STAT investigation reported that the FDA had failed to keep the public informed on its regulation of AI devices, or crucial details on the tests undertaken to prove their safety and effectiveness.
Reviewers from the FDA have given their blessing to the Pfizer/BioNTech COVID-19 vaccine ahead of a key meeting tomorrow – but the regulator noted that there are still uncertainties about whether the shot can stop the disease from spreading. The post FDA reviewers back Pfizer/BioNTech COVID-19 vaccine ahead of panel appeared first on.
Lewis, Senior Regulatory Device & Biologics Expert — We were preparing this blogpost about FDA’s draft guidance on “Remote Interactive Evaluations” when we learned something. Compared with the COVID-centered version of the document released in April 2021, there is very little that is new or different.
Lewis, Senior Regulatory Device & Biologics Expert — FDA recently published the final guidance document “ Comparability Protocols for Postapproval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA.” Brevig, Senior Regulatory Device and Biologics Exper & Richard A.
As a government shutdown looms, FDA published a contingency plan stating it will immediately have to furlough 19% of its staff until it receives more funds. Food and Drug Administration will have to furlough almost one-fifth of its staff and halt work that could have a critical impact on medical-products industries , Regulatory Focus says.
On February 7, at a town hall organised to discuss clinical trial designs for gene therapies, FDA experts pushed pharma players to look for ways to establish clinical effectiveness despite the challenges in recruiting patients with rare diseases.
Farquhar — Francis Godwin, Director of the Office of Manufacturing Quality of the Office of Compliance at FDA’s Center for Drug Evaluation and Research provided useful information (presentation attached here ) Tuesday at the GMP by the Sea Conference.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
There is a lot of discussion about the power of drawing out insights from information hidden within electronic health records and insurance claims, but little regulatory guidance on how that should be undertaken. The FDA stresses it is a preliminary document and is encouraging comments, which can be filed up to 60 days after publication.
Baumhardt, Senior Medical Device Regulation Expert — As an end of the year gift, FDA finalized its guidance document, Digital Health Technologies for Remote Data Acquisition in Clinical Investigations , in late December. c) a submission of an IDE to the FDA is not required. By Adrienne R.
Walsh — Last fall, we blogged about the process FDA uses to review allegations of regulatory misconduct against device manufacturers, suggesting greater transparency on the FDA process was needed (see here ). FDA also requests a “detailed description of the allegation with any available supporting documentation.”
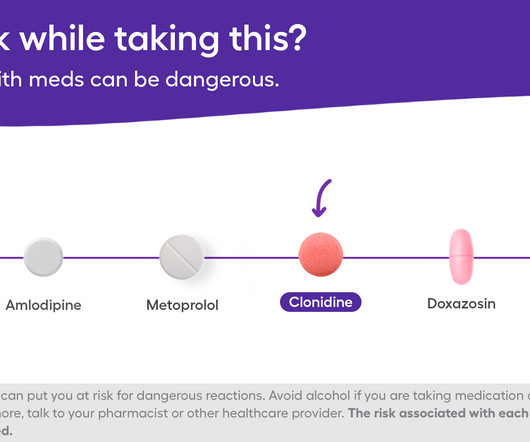
According to its prescribing information, it works to lower blood pressure by attaching to certain receptors (proteins) in the brain. In rare cases, clonidine can cause liver problems, such as hepatitis and increased liver enzymes, according to its prescribing information. Alcohols negative effects on the liver are well-documented.
In 2006, FDA stated that DMCs were not recommended “for most clinical studies,” particularly those in early product development, short-term studies, or studies addressing less severe outcomes. In another update, the recent draft guidance added “entities reviewing safety data” and adaptation committees.
Food and Drug Administration (FDA) regulation as a medical device. Determining whether a product is a medical device subject to FDA regulation necessarily begins with understanding the FDA regulatory definition of ‘medical device’. When assessing mHealth technology, the FDA will determine whether an application is either: i.
Last November, an employee at Intas Pharmaceuticals, which makes several widely used chemotherapies that are in short supply, was seen pouring acetic acid in a trash bin containing documents at a manufacturing facility. Another employee failed to report all test results on product samples and, at times, printouts were tossed in the trash.
Lenz, Principal Medical Device Regulation Expert — FDA recently issued a draft guidance which would update the agency’s Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions guidance. FDA provides examples of changes that are likely and unlikely to impact cybersecurity of an existing device.
Lenz, Principal Medical Device Regulation Expert — For medical device start-up companies, understanding and successfully navigating applicable FDA regulations and requirements is an important part of the path to market. There are many resources that provide information and guidance for each of these required elements.
On 27th September 2022, the Food and Drug Administration (FDA) issued its final guidance for industry and FDA staff clinical decision support (CDS) software, which has been anticipated since the Center for Devices and Radiological Health (CDRH) listed the guidance as a top priority for fiscal year 2022. Criteria for regulation.
Gibbs & Ana Loloei & Véronique Li, Senior Medical Device Regulation Expert — FDA has long touted the use of real-world evidence ( RWE ). FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices.
Memorizing 45+ page document is certainly not a reasonable expectation, but one can certainly walk away with an awareness of general concepts and themes which are relevant. ” Labeling the use of an antibiotic as inappropriate or appropriate cannot simply be done based upon whether it is FDA-approved for a given indication.
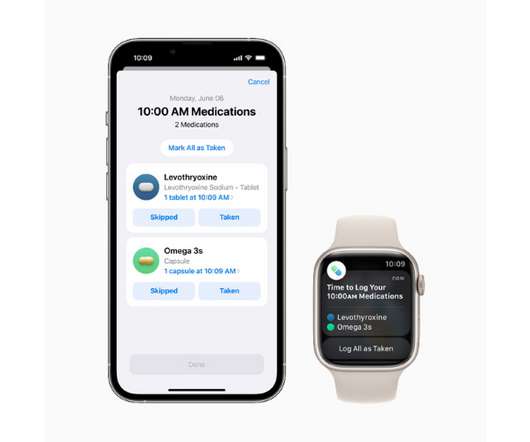
Other features include compatibility with Apple’s health data sharing facility, which allows information to be shared securely with healthcare staff or carers, and the ability to export a medications list into a PDF. For example, it can warn of possible issues like mixing drugs like metronidazole or cetirizine with alcohol.
Baumhardt, Senior Medical Device Regulation Expert — With the explosion of health‑related software, many software developers are generating products with functionality that is subject to regulation by the Food and Drug Administration (FDA). Please check out FDA’s presentation on this very topic – Is My Product a Medical Device?
It is approved by the Food and Drug Administration (FDA) for treating Type 2 diabetes. Mounjaro is not generally covered for weight loss since its not FDA approved for weight loss. Zepbound and Mounjaro are virtually the same drug with similar dosages but different FDA-approved uses.
Sponsors running oncology and diabetes trials, for example, will ask for more information on patients’ real-life movements to demonstrate their quality of life, as well as efficacy and safety.”. Teams will develop, review, approve, and track content and documents more closely than ever, right down to version history and user access.
Specifically, the change of ownership/location documents are quite a bit different and include some new and helpful information to assist applicants wading through the very confusing and cumbersome licensing process. The California Board of Pharmacy updated many of the application forms and guidelines in late 2024.
FDA , Petitioners, a liquid nicotine manufacturer, sued FDA arguing that the Agency was arbitrary and capricious in rejecting the Petitioner’s Premarket Tobacco Application (“PMTA”) in violation of the Administrative Procedure Act (“APA”). Youth behavioral data was not specifically required but FDA encouraged such information.
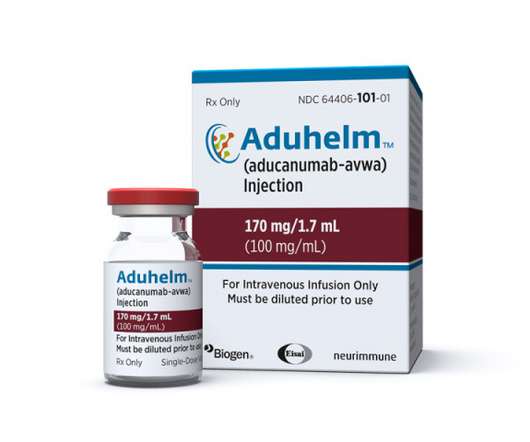
The FDA’s interactions with Biogen in the build-up to last year’s controversial approval of Alzheimer’s disease therapy Aduhelm have been described as “inappropriate” and “atypical” in a congressional report on the review.
Livornese — FDA issued a Federal Register notice on September 13, 2024, seeking feedback on the Integrated Review format that the Center for Drug Evaluation and Research (CDER) began using as part of its New Drugs Regulatory Program (NDRP) modernization effort several years ago. By Deborah L. 89 FR 74966 at 74968 (Sept. 13, 2024).
By Véronique Li, Senior Medical Device Regulation Expert — A joint eSTAR pilot (which we previewed in November) between FDA and Health Canada has now been launched. Responses from FDA and Health Canada to interested sponsors are expected within 3 business days of the email request. checks for incomplete sections.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. FDA further retained some definitions in the QSMR. The new § 820.10 Revised § 820.3
Walsh — In our June blog post , we reported on FDA’s request for comments about its program to receive information from the public alleging misconduct by other companies. FDA uses this program to help it identify risks and to determine whether further investigation is needed. 552), there are exemptions from mandatory disclosure.
Shapiro — Last summer, FDA published a draft guidance, Laser-Assisted In Situ Keratomileusis (LASIK) Lasers – Patient Labeling Recommendations (July 29, 2022) setting forth a proposal for new recommended patient‑directed labeling. The approved devices already have patient‑directed labeling that FDA has approved. By Jeffrey K.
Shapiro — More than a decade ago, FDA began systematically to incorporate review of human factors (HF) design validation within 510(k) reviews. Now FDA has issued a draft guidance , Content of Human Factors Information in Medical Device Marketing Submissions (Dec. This approach is one-size-fits-all.
Food and Drug Administration (FDA) for chronic weight management in adults with a starting, or baseline , body mass index ( BMI ) of 30 or more ( obesity ). You can contact your specific plan for more information about weight loss drug coverage , and specifically, Saxenda. It is approved by the U.S.
Javitt — FDA recently published a long-awaited draft guidance aimed at reducing the need for prior FDA authorization of modifications to artificial intelligence/machine learning (AI/ML)-enabled device software functions (ML-DSFs). a) , and related guidance documents (e.g., See 21 CFR 807.81(a)(3) a)(3) and 21 CFR 814.39(a)
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content