This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA advisors will scrutinise three cancer immunotherapies granted conditional approvals at a three-day meeting this week, to see if they should stay on the market. . Briefing documents published by the FDA ahead of the meeting suggest that discussion will focus on ongoing trials that may serve as alternative confirmatory studies.
Compounding pharmacies are typically allowed to make copies of drugs that are deemed to be in shortage by the FDA, which semaglutide has been for over two years. The FDA still has to make a decision on whether to officially place semaglutide on the lists. regulators in 2011. appeals court ruling in its favor , Reuters writes.
This is the third withdrawal of an accelerated approval FDA has performed, following Avastin in 2011 and Makena in 2023. Without commenting on the merits of the decision, it is an interesting window into how FDA may use these new procedures in practice. Tobolowsky & Michelle L.
Lenz, Principal Medical Device Regulation Expert — On September 6, 2023, FDA announced its latest efforts to modernize the 510(k) process, outlining FDA’s latest improvements to strengthen the 510(k) Program and announcing release of three draft guidance documents. stated, “ We appreciate the IOM’s report on the 510(k) program.
The application of Bayesian methodology has been recognised by the US Food and Drug Administration (FDA) as useful in early phase clinical trials involving paediatric populations. FDA presentation. 2011 May; 46(3): 399–424. FDA Rare Diseases: Natural History Studies for Drug Development Guidance for Industry. Patel et al.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
The case revolves around allegations that Novartis made an agreement in 2011 with Par Pharmaceutical when Exforge (valsartan/amlodipine) was nearing the end of its patent life that was designed to keep Par’s generic version of the drug off the market until 30 September, 2014.
This class-action litigation specifically focused on a 2011 licensing agreement between the two companies. According to the legal documents , the plaintiffs accused the companies of “violating federal antitrust laws, alleging “per se” and “rule of reason” violations”. Fighting Entresto generics.
Food and Drug Administration (FDA) certifies the Red Dye 40 as safe for human consumption. Apart from Red Dye 40, there are eight other certified color additives approved for use in food by the FDA: Blue No. The FDA has denied any link between the two for years. The controversy surrounding Red Dye 40 The U.S. 1, Blue No.
In 2011, following referral from another company, IMPACT (now part of Syner-G BioPharma Group) was contacted by a small development stage pharmaceutical company located in the United Kingdom to assist them with pre-IND activities for a new chemical entity for the treatment of asthma. Authored the client’s Pre-IND Briefing Document.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Q13 Development Timeline The process of drafting the new guidance document was initiated in earnest in November 2018 when the concept paper 2 and business plan were endorsed. Since then, agencies have begun officially adopting the guidance, with the FDA doing so in March 2023.
Although the pharmaceutical industry has consistently improved manufacturing processes 3 in compliance with good manufacturing practices, 4 there are documented deviations from good practices 5 including the continued falsification of medicines. Directive 2011/62/EU of the European Parliament and of the Council.” Published 2011.
The only FDA approved treatment for post-partum depression is a progesterone analogue that must be delivered over a 60-hour IV infusion and appears mildly efficacious. There have been a couple of small case series document LSD use in pregnancy and normal birth outcomes, although these are not high quality or conclusive by any means [23].
Also, determining whether an intrusion occurred and documenting that intrusion adds to the challenges of performing aseptic technique properly (and perhaps adds to the subjectivity of the current control strategies). Published January 2011. Consistent 1 US Food and Drug Administration Center for Drug Evaluation and Research.
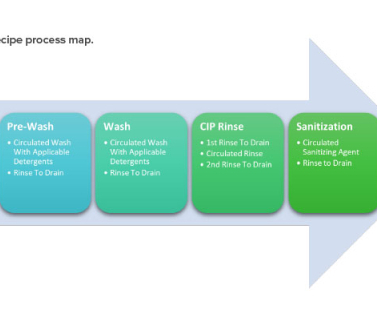
5 It includes cleaning validation master plan, product risk assessment, utility and equipment readiness, analytical method readiness, sampling site selection/grouping, standard operating procedures, validation protocols, execution of validation protocols, personnel training, and the validation documentation package. January 2011.
It builds on drug calculations topics previously described (1), including guidance documents (2–5). FDA guidance documents illustrate labeling salt drugs and associated active moieties. The secondary label will specify the Drug ZA-ion amount per capsule and the equivalent Drug Z-salt amount consistent with FDA labeling guidance.
Livornese — Last month, Congress took a big step towards improving clinical trial diversity by requiring sponsors of most drug and device clinical studies to submit a diversity action plan when they submit key trial documents to the Food and Drug Administration (FDA). This guidance was finalized in 2020. by the end of 2023).
government in the now-infamous (at least in FDA circles) Teva v. If an FDA-approved carve-out could support an intent to induce infringement claim, the use of the “section viii pathway would be substantially deterred.” Plainly, the Government brief states “The decision below is incorrect. GSK skinny label case , the U.S.
The first well-documented case of multiple myeloma was reported in 1844 by renowned British surgeon Samuel Solly. 2001– FDA green lights revolutionary treatments. Just over a decade after it was developed by biochemist Nicholas Lyndon, Imatinib received US Food and Drug Administration (FDA) approval in 2001.
This group also performed a randomized, larger placebo-controlled trial of 43 patients in 2011. All Lumebox products are FDA-registered, follow the Good Manufacturing Practice (GMP) system, and every batch is third-party tested. This Class II medical device is FDA-cleared and meant to target all kinds of pain. doi:10.1089/thy.2010.0102;
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. FDA CFR Title 21 Parts 211, 600, and 1271; 8. , FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice, 11.
FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. September 2022. 25%20from%202022%20to%202027 24 Eglovitch, J. 7 March 2023.
This article of mine discusses 19 thyroid-toxic products that have been banned by the FDA. Feel free to use my own health timeline as a guideline to document your health journey. Published September 12, 2011. The Environmental Working Group (EWG) also has consumer resources that can help individuals source cleaner options.
As of 2022, there were 15 drugs (see Table 1) manufactured using CM elements that have received FDA approval, with GSK, Pfizer, and Vertex owning approximately 60% of the market share, followed by Janssen/J&J with about 13%. Table 1: List of FDA-approved commercial products using CM elements.
In 1998, the Food and Drug Administration (FDA) defined an adaptogen as a new kind of metabolic regulator that has been proven to help in environmental adaptation and to prevent external harms. 2011 May;20(3):225-39. [11] Published 2019 Dec 9. doi:10.3390/molecules24244501 [10] Baliga MS, Dsouza JJ. Eur J Cancer Prev.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



























Let's personalize your content