This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Farquhar This is the first in a series of blog posts on tips for successfully handling an FDA inspection. Using publicly available examples, these lessons will illustrate potential pitfalls and strategies for interacting with FDA during and after an inspection. & Douglas B. FD&C Act 501(j).
Karst The FDA Reduction-in-Force (Termination)or RIF(T)announced last week has resulted in countless stories in the press and on personal LinkedIn accounts from those RIFd. As folks steeped in the world of generic drugs And Hatch-Waxman know, theres a lot that happens before FDA can take action on an ANDA.
Koblitz — Back in June, when Congress was negotiating the User Fee Acts, FDA asked Congress to add in some provisions reversing several lawsuits that it had just lost. Ultimately, FDA lost that fight, and a slimmed down version of the FDA Safety and Landmark Advancements (“FDASLA”) passed without those sections.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
Livornese As promised in the Fall Unified Regulatory Agenda, FDA issued the final rule to establish the pathway to obtain marketing approval of a nonprescription drug product with an additional condition for nonprescription use (ACNU) on December 26, 2024, before the end of the calendar year. By Deborah L. 105288 (Dec.
By Riëtte van Laack — On December 7, 2022, FDA announced the availability of the final guidance regarding the enforcement policy for homeopathic drug products. This concludes FDA’s reevaluation of the regulation of homeopathic drugs which it started in 2015. FDA denied that petition in 2019.
Food and Drug Administration (FDA) issued two guidance documents, one draft and one final, on food allergen labeling requirements. 1, 2023; The applicability of food allergen labeling requirements to specific products (e.g., 1, 2023; The applicability of food allergen labeling requirements to specific products (e.g.,
Michael Laposata and the Association for Molecular Pathology in the recent LDT litigation, the Federal, Food, Drug, and Cosmetic Act does not authorize FDA to regulate LDTs as medical devices. Just before this decisive event, though, FDA released a relatively rare Warning Letter to a manufacturer of research use only (RUO) reagents.
The Food and Drug Administration (FDA) plays a critical role in regulating the pharmaceutical industry and ensuring that medications and medical devices marketed to the public are safe, effective, and appropriately labeled. However, in recent years, there has been a noticeable decline in the number of these letters being issued.
Koblitz — Every year, federal agencies submit a budget request to Congress to fund various agency initiatives, and every year FDA includes a list of legislative proposals that it would like to see come out of Congress. FDA believes this change would effectuate timelier and more cost-efficient generic drug development.”
Shapiro — Last summer, FDA published a draft guidance, Laser-Assisted In Situ Keratomileusis (LASIK) Lasers – Patient Labeling Recommendations (July 29, 2022) setting forth a proposal for new recommended patient‑directed labeling. The approved devices already have patient‑directed labeling that FDA has approved.
Gaulkin & Riëtte van Laack — As readers of this blog know, there is a lot of contention about the naming of alternative protein products (APPs), including both plant-based and cell-cultured alternatives for (traditional) animal products. We’ve previously blogged about this ongoing battle here , here , here , and here. By Sophia R.
Baumhardt, Principal Medical Device Regulatory Expert FDA recently released its final guidance for Predetermined Change Control Plans (PCCPs) for Artificial Intelligence-Enabled Device Software Functions (AI-DSF). If FDA agrees to the PCCP, such changes can be made without a supplemental marketing submission.
Lenz, Principal Medical Device Regulation Expert — For several years, FDA has requested that sponsors of drug or biologic led combination products identify essential performance requirements (EPRs) related to the device constituent in their applications. By Adrienne R. does not use this term. does not use this term.
By Dara Katcher Levy — Yesterday, FDA published a new Draft Guidance, “ Communications from Firms to Health Care Providers Regarding Scientific Information on Unapproved Uses of Approved/Cleared Medical Products Questions and Answers ” (SIUU Guidance or Draft Guidance).
For both draft guidances, it is unclear what induced FDA to publish these draft guidances now. FDA indicates that this guidance is intended to help manufacturers ensure consistent content and format of the statement of identity and strength for all nonprescription drug products, which then will allow consumers to compare products.
Food and Drug Administration (FDA) released a draft update to its Compliance Policy Guide (CPG) for FDA staff on the Agency’s enforcement of major food allergen labeling and cross-contact. The draft CPG directs FDA field staff to examine possible food product adulteration due to labeling related to allergen cross-contact.
the ads can come from different sources and that can lead to run ins with the FDA. Accordingly, if a company is promoting a drug for catatonic schizophrenia, but the product is only approved for disorganized schizophrenia, such promotion could cause patients and clinicians to believe that the product is approved for an off label disease.
Livornese On Friday, March 21, 2025, FDA announced that it was further delaying the effective date for the ACNU final rule until May 27, 2025. FDA then delayed the effective date. By Deborah L. We wrote about the December 26, 2024 publication of the final rule (89 FR 105288) here.
” Labeling the use of an antibiotic as inappropriate or appropriate cannot simply be done based upon whether it is FDA-approved for a given indication. This blog post I did may also be of interest, on the topic. Daptomycin: from the mountain to the clinic, with essential help from Francis Tally, MD Find it here.
Hosted by American Conference Institute, the FDA Boot Camp returns for its 40th iteration with the continued intent of providing an essential working knowledge of core FDA concepts, and real-world examples that will help you to excel in your everyday practices. Labeling in the drug and biologics approval process.
After a firm submits a 510(k) to FDA, FDA will request still more information after a first-pass review. According to the 2 nd Quarter FY2023 MDUFA V Performance Report , FDA issued a request for additional information (AI request) on the first FDA review cycle for 63% to 68% of 510(k)s submitted in FY2018 to FY2022.
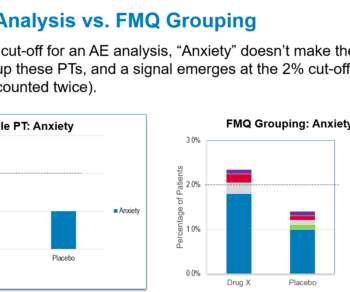
By Ellis Unger, Principal Drug Regulatory Expert — On September 14, 2022, FDA/CDER/Office of New Drugs, in collaboration with the Duke-Margolis Center for Health Policy, hosted a virtual meeting on advancing premarket safety analytics , including sessions on new FDA Medical Queries and standardized presentations of safety data.
Since that time, FDA issued a draft guidance for predetermined change control plans (PCCPs) for Artificial Intelligence/Machine Learning (AI/ML) software functions. See our prior blog post on the topic here.
Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. FDA further retained some definitions in the QSMR. Notably, Part 820 will look different. Revised § 820.3
FDA-2023-P-0313 and FDA-2023-P-0344 ) regarding its product Hetlioz (tasimelteon). Vanda requested that FDA revoke the approval of Apotex’s and Teva’s generic versions of Hetlioz on the grounds that the generic tasimelteon products did not meet the statutory “same labeling” requirement for generic drugs found in 21 U.S.C. §
By doing so, FDA has limited the number of tests that have reached the market, thereby reducing available supply and increasing prices. As FDA would acknowledge, the antigen tests are the fastest and most practical method for distributing testing in the general population. That could happen again.
Walsh — Last fall, we blogged about the process FDA uses to review allegations of regulatory misconduct against device manufacturers, suggesting greater transparency on the FDA process was needed (see here ). By Véronique Li, Senior Medical Device Regulation Expert & Anne K. This statistic is quite disheartening.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. However, FDA’s legal standing to insist on cybersecurity features, especially within the substantial equivalence paradigm, has been questionable.
See our prior blog post summarizing the different phaseout stages and categories of enforcement discretion. Instead, we focus here on the few notable statements that provide new or more detailed guidance than FDA has previously offered.
Lenz, Principal Medical Device Regulation Expert — FDA recently released a new eSTAR template for device pre-submissions and 513(g) Requests for Information, referred to as PreSTAR. A pre-submission provides the submitter an opportunity to obtain FDA feedback prior to a planned medical device premarket submission.
The Court remanded to USDA, the BE labeling regulation provisions regarding these disclosure options without vacatur; in other words, the status quo is maintained (products using these disclosure options need not be revised immediately) while USDA revisits these two disclosure provisions of the BE labeling rule.
Livornese — After teasing a new rapid review pilot program for the past few weeks, on June 17, 2025, FDA officially announced the Commissioner’s National Priority Voucher (“CNPV”) program to expedite new drug and biologic (but not device or drug-device combination product) reviews. Tobolowsky & Michelle L. Butler & Deborah L.
Here I share some of the latest news, articles, editorials, or blog posts that fall generally under the theme of Pharma or healthcare. NEW FDA WARNING: OPIOIDS MAY INCREASE PAIN! I might throw in a something off-topic from time to time which I found while wandering throughout the endless hallways and corridors of the internet.
Designed for in-house legal and compliance counsel, industry executives, and private practice attorneys working for the OTC drug industry, the event will welcome distinguished industry thought leaders – including from the National Advertising Division and FDA – to share their expertise and strategic insights.
Loloei is a 14-year veteran of the FDA, where most recently she served as a Senior Regulatory Counsel in the Office of Policy at CDRH. While at FDA, Ms. During her FDA tenure, Ms. It was a true honor and privilege to collaborate with so many fantastic coworkers and partners for nearly 14 incredible years at FDA.
By Ricardo Carvajal — FDA announced that it has completed the first pre-market consultation for an animal cell-cultured food, based on a safety assessment submitted by UPSIDE Foods. FDA’s response letter to UPSIDE Foods indicates that the safety standard applied by the company was one of comparable safety (i.e.,
2022, FDA published a draft guidance on FDA’s implementation of the Over-the-Counter Monograph Drug User Fee Program (OMUFA). Much of the guidance repeats the law and FDA’s previous notices (see e.g., our blog posts here and here ). FDA is authorized to charge an annual facility fee for OTC monograph drug facilities.
FDA Law Blog readers receive a 10% discount off the tuition fee (promo code D10-999-FDA25 ). s John W.M. Claud will be speaking at the conference in a session titled “Crafting Your Safety Blueprint for Adverse Events and Recalls under MoCRA.” You can register for the conference here.
The American Conference Institute is hosting their 39th FDA Boot Camp from September 14-15, 2022. It is important for attorneys who do not have regulatory practices and life sciences executives who deal with FDA-regulated products to have a familiarity with these concepts. The conference will be held virtually.
Promotional labeling is generally any labeling other than FDA-required labeling that is devised for the promotion of a product, as well as other functions, and can include printed, audio, or visual matter that describes the product. l)(1) (e.g.,
Javitt — FDA recently published a long-awaited draft guidance aimed at reducing the need for prior FDA authorization of modifications to artificial intelligence/machine learning (AI/ML)-enabled device software functions (ML-DSFs). Baumhardt, Senior Medical Device Regulation Expert & Philip Won & Gail H. See 21 CFR 807.81(a)(3)
Walsh — The question of whether FDA has authority to take pictures during an inspection of a facility has bounced around the food and drug bar for many years. FDA’s view is stated in the agency’s Investigation Operations Manual, and it is one with which a number of practitioners disagree. By Ricardo Carvajal & Anne K.
Shapiro — More than a decade ago, FDA began systematically to incorporate review of human factors (HF) design validation within 510(k) reviews. Now FDA has issued a draft guidance , Content of Human Factors Information in Medical Device Marketing Submissions (Dec. FDA did not require HF data as a basis for clearance of the predicate.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content