This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
First published in 1989, there have been a total of 5 adaptations in 1996, 2003, 2005, 2007 and 2009, but no complete revision. Annex 1 of the EC GMP Guide " Manufacture of Sterile Medicinal Products " has a long history. In 2012, there was a proposal for a complete revision which resulted in a concept paper in 2015.
Bob also initiated an agreement under which ISPE’s Guidance Documents are made available to PIC/S and WHO inspectors, which is still in place today. Most recently he assisted the regulatory authorities of Saudi Arabia, Russia, China, Jordan, and Philippines in the process to attain PIC/S membership.
In the US, botanical dietary supplements can be sold without US Food and Drug Administration (FDA) approval, 1 prompting many vendors to promote the use of botanical products as dietary supplements , 2 rather than pursue a path of regulatory approval. 3 Is two too few? 3 Is two too few?
To give examples, such a work package focuses on the identification of regulations (ICH Q2 R2, 3 Q8-Q14, 4-10 FDA PAT guidance, 11 EMA Annex 15 12 ), some of which may be mainly guidance documents with recommendations and flexibility when it comes to PAT.
Lenz, Principal Medical Device Regulation Expert — Since 2005, 510(k) submissions have been formatted according to FDA guidance Format for Traditional and Abbreviated 510(k)s. Several sections of the eSTAR templates have questions that walk through related guidance documents. By Adrienne R. Know Your Guidances.
It is estimated that only one in 10 drugs that enter Phase I trials are subsequently licensed by the US Food and Drug Administration (FDA). 5 When these medicines are prescribed to patients, we can use feedback from the real-world data that documents clinical outcomes, efficacy measures, patient-reported outcomes, and adverse events.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. Additional required information beyond ICH guidelines, or non-value-added documents (e.g., 6, and 2.3.S.7 7 are required.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. Published 15 November 2021.
Concept Paper: M4Q(R2) Common Technical Document on Quality Guideline.” As part of the US Food and Drug Administration (FDA) QbD pilot 4 in 2005–2006, the need to convey how the control strategy is linked to the target product profile (TPP) and quality target product profile (QTPP) was discussed. Published 15 November 2021.
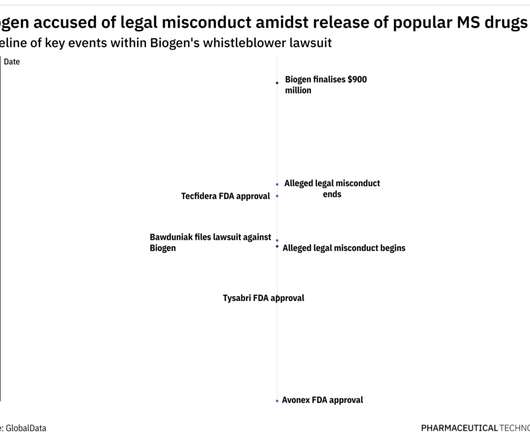
These, along with other similar claims, meant Biogen owed potential damages of $1,036,900,151 to the US and the various States, as per court documents. In 2005, Biogen withdrew Tysabri from the market following a clinical trial that resulted in two participants contracting progressive multifocal leukoencephalopathy (PML).
Also, determining whether an intrusion occurred and documenting that intrusion adds to the challenges of performing aseptic technique properly (and perhaps adds to the subjectivity of the current control strategies). November 2005. Vials are rejected or discarded when critical zone intrusion is detected during operator interventions.
Why, then, are FDA inspections abroad so infrequent and disappointing? FDA Inspections Suspended: During the COVID pandemic, the Food and Drug Administration suspended its inspections at most foreign pharmaceutical facilities. Even under the best of conditions, the FDA never inspects all foreign pharmaceutical plants annually.
It builds on drug calculations topics previously described (1), including guidance documents (2–5). FDA guidance documents illustrate labeling salt drugs and associated active moieties. The secondary label will specify the Drug ZA-ion amount per capsule and the equivalent Drug Z-salt amount consistent with FDA labeling guidance.
Recent US FDA data show that the lack of raw material availability contributes to 27% of drug shortages (see Appendix ). A formal change control system under the company’s pharmaceutical quality system (PQS) is required to evaluate all raw material changes, with established procedures for identification, documentation, review, and approval.
For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. FDA CFR Title 21 Parts 211, 600, and 1271; 8. , FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice, 11.
2 (1 May 2005). FDA Official Warns Manufacturers of Common Problems Found in Aseptic Operations.” www.raps.org/news-and-articles/news-articles/2023/3/fda-official-warns-manufacturers-of-common-problem 25 US Food and Drug Administration Center for Drug Evaluation and Research. September 2004. link] 19 Agalloco, J., 20 Parrish, M.
Quality by design (QbD) is a concept introduced into the pharmaceutical regulatory lexicon in 2005. QbD principles are woven into regulatory guidance documents, primarily guidances Q8 to Q11 of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
Livornese & JP Ellison — On November 8, 2024, FDA issued a proposed order to remove the oral decongestant ingredient phenylephrine (including both phenylephrine hydrochloride and phenylephrine bitartrate) (collectively, PE) from the OTC monograph on the basis of a lack of effectiveness. By Deborah L. a); OTC Monograph M012: § M012.20.
A few years ago, I had a few clients with documented elevations in prolactin, and I have seen that lowering prolactin levels can help with reducing thyroid antibodies. Studies have, however, documented that bromocriptine, a medication used to lower prolactin levels, can reduce flares in lupus, another autoimmune condition.
In 1998, the Food and Drug Administration (FDA) defined an adaptogen as a new kind of metabolic regulator that has been proven to help in environmental adaptation and to prevent external harms. Epub 2005 Oct 7; Costa CA, Tanimoto A, Quaglio AE, et al. Int J Ayurveda Res. 2010;1(1):37-40. 2006 Feb 16;78(12):1287-92.
We organize all of the trending information in your field so you don't have to. Join 5,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





















Let's personalize your content