New workflow for the analysis of mRNA therapeutics
Posted: 1 March 2023 | Mark Dickman (University of Sheffield) | No comments yet
Here, Mark Dickman, Professor in Bioanalytical Science and Engineering at the University of Sheffield, explores how analytical techniques such as liquid chromatography-mass spectrometry can support the manufacturing of mRNA therapeutics.

Large RNA is rapidly emerging as a new class of therapeutics. Nowhere is this more publicly evident than in the development, regulatory approval, and efficacious use of mRNA‑based vaccines for COVID-19. Beyond the high-profile COVID-19 vaccines, mRNA therapeutics are crucial in applications such as protein replacement therapies and regenerative medicine. Scaling up the production of mRNA therapeutics to meet today’s growing demand requires improved optimisation of the associated analytics. An automated liquid chromatography‑mass spectrometry (LC-MS) workflow used in conjunction with RNase mass mapping is proving successful in simplifying and accelerating mRNA analysis.
mRNA therapeutics and their manufacture
mRNA therapeutics contain a specific sequence (coding sequence), which corresponds to the gene or protein of interest. The mRNA is packaged and delivered to the cytoplasm of host cells, where it is translated into a protein that exerts the desired cellular effect. This mRNA is formulated within a delivery vehicle, usually lipid nanoparticles, and once inside the target cell, it “hijacks” the cellular machinery to make the desired protein.
Manufacturing mRNA for therapeutic use involves in vitro transcription (IVT). This enzymatic process employs a DNA template, typically plasmid DNA, which carries the mRNA sequence of interest. After the DNA template is linearised, the addition of T7 RNA polymerase and nucleoside triphosphate (NTP) building blocks enable enzymatic production of the mRNA containing the required protein-coding sequence.
While the manufacturing process is relatively simple, it results in the generation of numerous contaminants and unwanted by-products. These include T7 RNA polymerase, NTPs, plasmid DNA, double-stranded RNA (dsRNA), failure sequences, degradation products, incomplete capping and polyadenylated products. All present a significant challenge from an analytical perspective for the downstream purification and analysis of mRNA therapeutics.
The urgent need for improved analytics
Current analytical methods for characterising RNA therapeutics are limited and the development of strategies for large RNA (typically >1000 nucleotides), including mRNA vaccines, has proven especially challenging – from the separation processes involved to analysis of degradation products. Consequently, there is a need for faster, more robust, and higher-resolution analytics since these are critical for successful manufacturing development.
Global demand for vaccines is growing and there is increasing pressure to scale up production. Process optimisation depends on good analytics, which are essential to ensure consistent batch‑to-batch manufacturing, process repeatability, and quality of the mRNA produced. Monitoring critical quality attributes (CQAs) such as identity, contaminants, yield, and potential modifications is vital. Validated analytical methods also have a fundamental role in supporting clinical development and regulatory submissions.
RNase mass mapping and its challenges
Mass spectrometry (MS)-based methods offer a powerful approach for RNA analysis. MS-based RNase mapping methods have proven successful in various applications, including sequence mapping and identifying post-transcriptional modifications. Recent work applying this technology to large RNA has driven the development of a rapid automated method for direct characterisation and comprehensive sequence mapping of mRNA therapeutics.
Following the purification of the resulting mRNA, ion-pair reversed-phase high-performance liquid chromatography (IP-RP-HPLC) provides data on yield, quantity, and quality, but offers no sequencing information. Alternative tools are therefore needed for further analysis, and it is here that mass mapping is being applied.
RNase mass mapping is similar to protein and peptide mass fingerprinting. It involves digesting the mRNA of interest with a specific RNase (eg, RNase A or T1) to generate a series of oligonucleotides.”
RNase mass mapping is similar to protein and peptide mass fingerprinting. It involves digesting the mRNA of interest with a specific RNase (eg, RNase A or T1) to generate a series of oligonucleotides. Mass spectrometry, incorporating high mass accuracy and tandem MS analysis, is then applied to sequence the fragments.
This mapping technique is already being used to analyse RNA in processes such as CRISPR1 but applying it to much larger mRNA therapeutics is less straightforward. The smaller RNA molecules (~100 nucleotides) involved in CRISPR produce a limited number of different sequences following RNase digestion, making analysis simple. Conversely, mRNA therapeutics are thousands of nucleotides in length. Using high-frequency RNases, such as RNase A or T1, to digest them produces many identical short oligonucleotide sequences that map to multiple points on the mRNA.
Another significant issue has been the lack of robust software for oligonucleotide characterisation. Manual data interpretation is often used but not only is this laborious and time consuming, it also restricts experimental scope and limits both productivity and confidence in the results, especially where very large numbers of fragments are generated. A different approach is essential to overcome the challenges of mapping large RNA.
Implementing an effective automated workflow
The newly developed workflow2 involves the partial digestion of the mRNA therapeutic using RNase T1 immobilised on magnetic beads. This is followed by purification, sequencing, and automated data analysis.
Compared with standard RNase digestion processes, the controlled partial digestion using immobilised enzyme generates increased numbers of larger sized fragments. Unlike the smaller fragments resulting from a standard digestion of mRNA, these larger fragments have unique sequences that map to specific points on the mRNA molecule. Enzyme immobilisation also avoids RNase contamination of the chromatography column used in the separation step, thus preventing any further digestion.
High-resolution separation of the oligonucleotide fragments is critical to the success of subsequent analysis. In this workflow, separation is achieved in around 40 minutes with IP-RP-HPLC using triethylamine (TEA) in combination with hexafluoroisopropanol (HFIP) and an acetonitrile gradient during the elution. This typically results in sharp elution peaks and length‑based separation of a large number of oligonucleotide fragments.
MS analysis of the mRNA digest requires high-resolution accurate-mass (HRAM) capabilities.2 Following the application of MS, the final, and crucial, step of the workflow is the automated data analysis. The software used here enables automated identification of the oligonucleotide fragments and supports specific RNase mass mapping approaches, as illustrated in Figure 1. Its sequence manager allows the input of the sequence of interest together with any modifications.
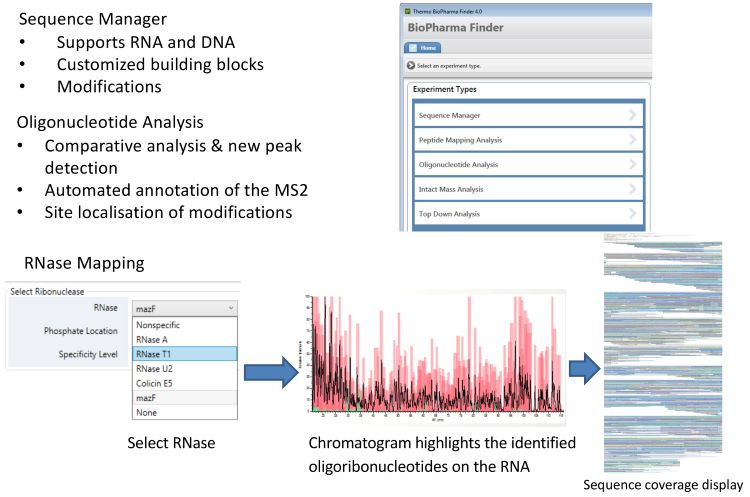
Figure 1: Automating data analysis.
The RNase mass mapping tool then generates a theoretical in silico digest and identifies oligonucleotide fragments based on the intact mass from high‑resolution MS1 spectra and the nucleic acid sequence and modifications from the MS2 spectra. The output chromatogram highlights the identified oligonucleotides on the RNA. This is then converted to a sequence map showing all the sequences identified and their locations on the mRNA sequence of interest.
Applying the workflow to mRNA therapeutics
Applying the developed workflow to mRNA therapeutics [SARS-CoV-2 spike protein mRNA and enhanced green fluorescent protein (eGFP) mRNA] confirmed that immobilising RNase T1 on magnetic beads gave tight control of the digestion process, generating highly reproducible partial T1 digests and separations. With respect to sequence mapping, Figure 2 shows the sequence coverage data in experiments performed using partial RNase T1 digests and traditional complete RNase T1 digests for both SARS-CoV-2 spike protein mRNA and eGFP mRNA. Using partial digests, approximately 85 percent sequence coverage was achieved. In contrast, complete RNase T1 digests gave many non-unique fragments, resulting in only 10-25 percent sequence coverage. As a control against false positive identifications, a random sequence of the same size and same GC content as the target mRNAs was included in all experiments. Searching the MS data against this random sequence gave extremely low sequence coverage, confirming the quality of the data.
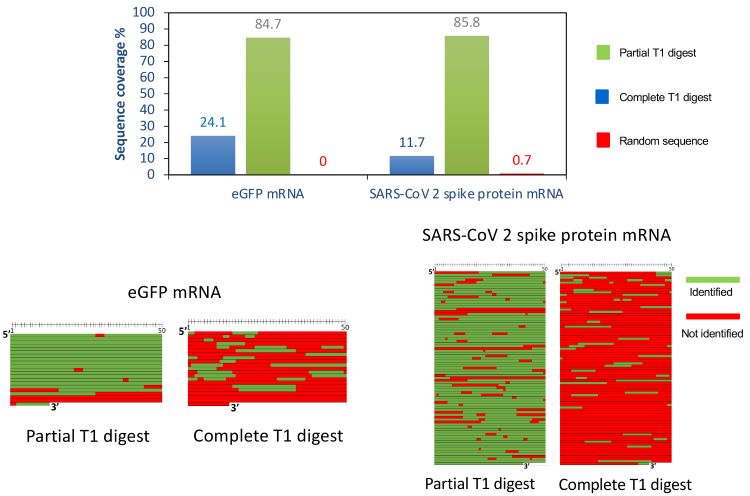
Figure 2: Sequence mapping of mRNA therapeutics.
Building confidence in results
Also important is the ability of the software to identify different sequence isomers. Even using the partial digestion strategy, several isobaric oligonucleotides with the same nucleic acid composition can be generated, each of which corresponds to a different sequence. Differential separation using IP-RP‑HPLC in conjunction with the tandem MS, followed by automated software analysis, enables clear identification of the different sequences.
Further confidence in sequence identification is gained from IP-RP-HPLC conditions generating several different charge states for each oligonucleotide fraction. Tandem MS then delivers sequence information on each of these oligonucleotide fractions.
A method fit for purpose
Mass spectrometry techniques are widely used for RNA analysis. Unfortunately, the large molecular size of mRNA presents challenges in methods such as RNase mass mapping, where the use of high frequency RNases fails to generate unique sequences for analysis. Reliable characterisation is, however, critical to successful manufacturing scale-up for the growing body of mRNA therapeutics in production and in the pipeline. New approaches are therefore needed.
Mass spectrometry techniques are widely used for RNA analysis.”
The LC-MS workflow described delivers high-quality results while simplifying and automating the process. It uses RNase T1 immobilised on magnetic beads to achieve a controlled partial digestion of the mRNA. This results in fragments that are larger and unique in sequence than those generated using standard RNase mapping methods. The partial digest is separated by IP‑RP-HPLC and analysed by HRAM MS. The data is automatically processed to generate high confidence identification of oligonucleotide fragments and comprehensive mRNA sequence mapping, which avoids the need for laborious manual interpretations and gives high confidence in the results.
Overall, the new workflow delivers rapid characterisation of mRNA therapeutics. It provides the information needed for identity testing, sequence validation, and analysis of impurities, essential for successful manufacturing and scale-up.
About the author
 Dr Mark J Dickman, PhD is currently a Professor in Bioanalytical Science and Engineering in the Department of Chemical and Biological Engineering at the University of Sheffield. He obtained a first-class honours degree in biochemistry/chemistry at the University of Sheffield. Mark continued at the Krebs Institute, where he obtained his PhD. Later, he joined a biotechnology company, Transgenomic Ltd, where he worked as a research scientist developing analytical techniques, including DNA/RNA chromatography.
Dr Mark J Dickman, PhD is currently a Professor in Bioanalytical Science and Engineering in the Department of Chemical and Biological Engineering at the University of Sheffield. He obtained a first-class honours degree in biochemistry/chemistry at the University of Sheffield. Mark continued at the Krebs Institute, where he obtained his PhD. Later, he joined a biotechnology company, Transgenomic Ltd, where he worked as a research scientist developing analytical techniques, including DNA/RNA chromatography.
References
1. Tamulaitis G, Kazlauskiene M, Manakova E, et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell. 2014 Nov 20; 56(4), 506‑17. https://pubmed.ncbi.nlm.nih.gov/25458845/
2. Vanhinsbergh CJ, Criscuolo A, Sutton JN, et al. Characterization and Sequence Mapping of Large RNA and mRNA Therapeutics Using Mass Spectrometry. Anal. Chem. 2022; 94 (20), 7339-7349 https://pubs.acs.org/doi/10.1021/acs.analchem.2c00765
Issue
Related topics
Analytical techniques, Biologics, Drug Development, Drug Manufacturing, Manufacturing, mRNA, Research & Development (R&D), Technology, Therapeutics, Vaccines, Viruses